Today, I review, link to and excerpt from Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup [PubMed Abstract] [Full-Text HTML] [Full-Text PDF]. Alzheimers Dement. 2024 Jun 27. doi: 10.1002/alz.13859. Online ahead of print. Clifford R. Jack Jr., J. Scott Andrews, Thomas G. Beach, Teresa Buracchio, Billy Dunn, Ana Graf, Oskar Hansson, Carole Ho, William Jagust, Eric McDade, Jose Luis Molinuevo, Ozioma C. Okonkwo, Luca Pani, Michael S. Rafii, Philip Scheltens, Eric Siemers, Heather M. Snyder, Reisa Sperling, Charlotte E. Teunissen, Maria C. Carrillo.
All that follows is from the above resource.
Abstract
The National Institute on Aging and the Alzheimer’s Association convened three separate work groups in 2011 and single work groups in 2012 and 2018 to create recommendations for the diagnosis and characterization of Alzheimer’s disease (AD). The present document updates the 2018 research framework in response to several recent developments. Defining diseases biologically, rather than based on syndromic presentation, has long been standard in many areas of medicine (e.g., oncology), and is becoming a unifying concept common to all neurodegenerative diseases, not just AD. The present document is consistent with this principle. Our intent is to present objective criteria for diagnosis and staging AD, incorporating recent advances in biomarkers, to serve as a bridge between research and clinical care. These criteria are not intended to provide step-by-step clinical practice guidelines for clinical workflow or specific treatment protocols, but rather serve as general principles to inform diagnosis and staging of AD that reflect current science. Highlights
- We define Alzheimer’s disease (AD) to be a biological process that begins with the appearance of AD neuropathologic change (ADNPC) while people are asymptomatic. Progression of the neuropathologic burden leads to the later appearance and progression of clinical symptoms.
- Early-changing Core 1 biomarkers (amyloid positron emission tomography [PET], approved cerebrospinal fluid biomarkers, and accurate plasma biomarkers [especially phosphorylated tau 217]) map onto either the amyloid beta or AD tauopathy pathway; however, these reflect the presence of ADNPC more generally (i.e., both neuritic plaques and tangles).
- An abnormal Core 1 biomarker result is sufficient to establish a diagnosis of AD and to inform clinical decision making throughout the disease continuum.
- Later-changing Core 2 biomarkers (biofluid and tau PET) can provide prognostic information, and when abnormal, will increase confidence that AD is contributing to symptoms.
- An integrated biological and clinical staging scheme is described that accommodates the fact that common copathologies, cognitive reserve, and resistance may modify relationships between clinical and biological AD stages.
1 BACKGROUND
Although the purpose of this document is to update the 2018 document, a set of fundamental principles emerged from prior NIA-AA workgroups. These principles, outlined in Box 1, are carried forward and serve as the foundation or starting point for the current revised criteria.
BOX 1: Fundamental principles
It is necessary to separate syndrome (clinically identified impairment) from biology (etiology).
Alzheimer’s disease (AD) is defined by its biology with the following implications.
AD is defined by its unique neuropathologic findings; therefore, detection of AD neuropathologic change by biomarkers is equivalent to diagnosing the disease.
AD exists on a continuum. The disease is first evident in vivo with the appearance of disease-specific Core biomarkers while people are asymptomatic. Pathophysiologic mechanisms involved with processing and clearance of protein fragments may be involved very early in the disease process, but these are not yet well understood.
Symptoms are a result of the disease process and are not necessary to diagnose AD.
Unimpaired individuals with abnormal biomarker test results are at risk for symptoms due to AD. They are not at risk for a disease they already have.
Clinical syndromes commonly seen with AD may also be caused by disorders other than AD, and therefore clinical presentation alone is not diagnostic of AD.
The same AD biology may result in different phenotypic presentations.
Three major developments prompted this update. First, treatments that target core disease pathology have, for the first time, received regulatory approval. The prospect of these therapies entering clinical practice highlights the importance of conceptual alignment among clinicians, industry, and academia around diagnosis and staging of AD.
Second, the most significant advance in AD diagnostics in recent years has been the development of blood-based markers (BBM) with some (not all) assays exhibiting accurate diagnostic performance. This now makes the biological diagnosis of AD (which previously required positron emission tomography [PET] or cerebrospinal fluid [CSF] assays) more generally accessible and is projected to revolutionize clinical care and research. The field is now in a transition phase during which BBM are being integrated with traditional CSF and PET biomarkers.
Finally, an important product of recent research is the recognition that imaging, CSF, and BBM within a pathobiological AT(N) (amyloid/tau/neurodegeneration) category are interchangeable for some, but not all, intended uses. The present document is updated to reflect this.
This is a forward-looking document based on current scientific evidence that provides a common framework for AD diagnostic and staging criteria to inform both research and clinical care. We do not provide detailed guidance on clinical workflow or treatment protocols; formal clinical practice guidelines will appear in a subsequent document. The criteria we describe are presently operationalizable at some but by no means all centers even among major medical institutions in high-income countries. We therefore view these criteria as a bridge between research and clinical care.
2 BIOMARKER CATEGORIZATION
Categorization of biomarkers refers to grouping biomarkers into categories that reflect a common proteinopathy pathway or pathogenic process. Categorization of biomarkers in the 2018 NIA-AA framework assumed equivalence of CSF and imaging biomarkers within each AT(N) category.8 Ample evidence has accumulated that this is not always the case; therefore, in these revised criteria we break from the assumption of equivalence between imaging and biofluid biomarkers within a given biomarker category.
We group biomarkers into three broad categories: core biomarkers of AD neuropathologic change (ADNPC),5 non-specific biomarkers that are important in AD pathogenesis but are also involved in other brain diseases, and biomarkers of common non-AD copathologies (Table 1). Within each of these three broad categories we subcategorize biomarkers by the specific proteinopathy pathway or pathogenic process that each measures; for example, “A” biomarkers denote the amyloid beta (Aβ) proteinopathy pathway.
Throughout the document we distinguish between imaging and fluid biomarkers. Imaging biomarkers measure cumulative effects; capture topographic information; map onto established neuropathologic constructs; and, in the case of amyloid and tau PET, represent insoluble aggregates.9–15 Fluid biomarkers reflect the net of rates of production/clearance of analytes at a given point in time.
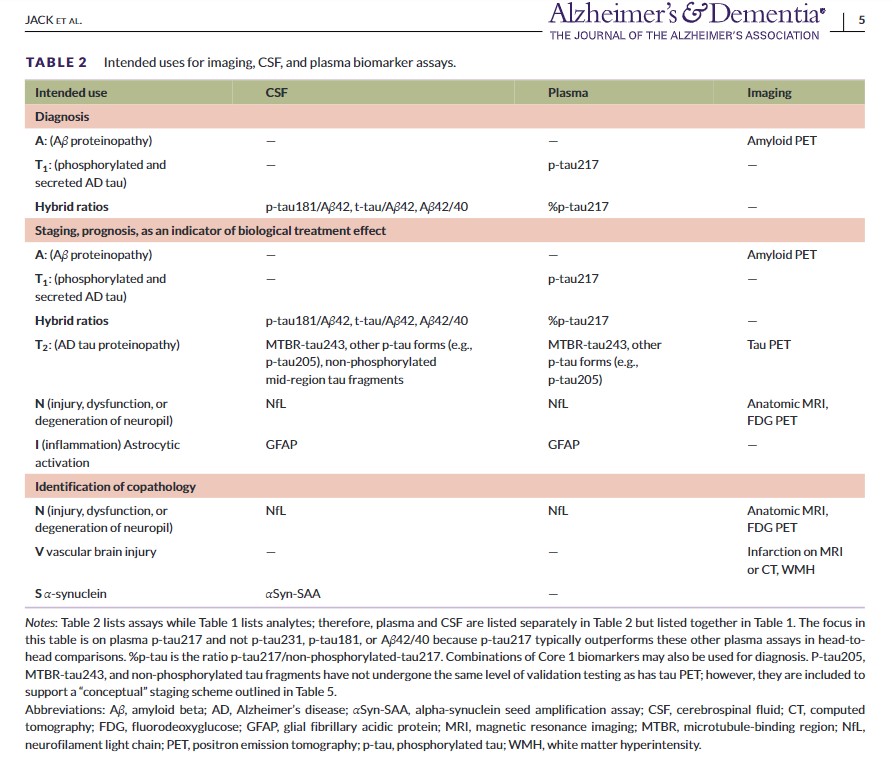
The 2018 framework recognized the need to modify the AT(N) biomarker classification scheme to incorporate newly developed biomarkers within an existing AT(N) category; and, we have included recently developed BBM of A, T, and (N) in this update. The 2018 framework also called for incorporating new biomarker categories beyond AT(N) as appropriate. This was denoted in the 2018 document as ATX(N), where X indicated a new biomarker category beyond A, T, or (N).7 Accordingly, Tables 1 and 2 include three new biomarker categories: inflammatory/immune mechanisms (I), vascular brain injury (V), and alpha-synucleinopathy (S). V and S biomarkers are relevant to this document on AD diagnosis and staging because AD most often occurs with copathologies in older adults.
Table 1 illustrates biomarker categories by pathogenic mechanism or proteinopathy pathway. CSF and plasma are listed together as fluid analytes because the same analyte is measured in CSF or plasma. Table 2 lists intended uses for biomarkers, which fall into several categories: diagnosis; staging, prognosis, as an indicator of biological treatment effect; and identification of copathologies. While Table 1 lists fluid analytes, Table 2 lists assays and, accordingly, CSF and plasma are broken into separate columns in Table 2 because assay implementation may differ between CSF and plasma. Table 2 also includes hybrid ratios, which are assays rather than individual analytes. Assays in Table 2 may be in vitro diagnostics, laboratory-developed tests, or research-use-only tests. The committee used the following criteria for inclusion in Table 2: the imaging, CSF, or BBM has either received regulatory approval or has played a prominent role in recent clinical research, and, in the opinion of the committee, enough evidence exists to support its clinical value and the assumption that it may receive regulatory approval in the future.
Tables 1 and 2 categorize core and non-core biomarkers. In the remainder of Section 2, we focus on core biomarkers of ADNPC to create a logical progression to the subsequent topics of diagnosis and staging, which use only core biomarkers. Non-core biomarkers (i.e., N, I, V, and S) are discussed in Section 7.
Core AD biomarkers are those in the A (Aβ) and T (tau) categories (Table 1). The A category denotes biomarkers of the Aβ proteinopathy pathway. Soluble aggregation-prone Aβ peptides are the molecular building blocks of insoluble fibrillar Aβ aggregates in plaques. Hence, fluid and imaging A biomarkers “represent different biochemical pools of the same proteinopathy pathway.”16 Moreover, although fluid Aβ42-based assays may become abnormal slightly before amyloid PET,17 the two are usually highly concordant.18–20
Timing relationships are different across the spectrum of T biomarkers. Phosphorylated mid-region fragments (phosphorylated tau [p-tau] 181, 217, and 231) become abnormal around the same time as amyloid PET and before tau PET.21–24 This has led to the suggestion that secretion of tau fragments phosphorylated at specific residues (181, 217, and 231) may represent a physiologic reaction to Aβ plaques25 and may link Aβ proteinopathy to early tau proteinopathy.26, 27 In contrast, other tau fragment analytes (e.g., microtubule-binding region [MTBR-tau243] and non-phosphorylated mid-region tau fragments) become abnormal later and correlate better with tau PET than with amyloid PET.28, 29 These observations led us to split the T biomarker category into two subcategories: T1 (biofluid analytes of soluble tau fragments that may reflect a reaction to amyloid plaques or to soluble Aβ species in plaque penumbra) and T2 (tau PET imaging or biofluid analytes that signal the presence of AD tau aggregates). Because of their time of onset, plasma p-tau 217, 181, and 231 have been proposed as biomarkers of Aβ plaques, but this is difficult to accept conceptually because these are tau fragments, not measures of the Aβ proteinopathy pathway. Furthermore, these tau analytes do correlate with tau proteinopathy in addition to Aβ proteinopathy.30 The T1 and T2 categories address these conceptual issues.
We introduce the concept of Core 1 and Core 2 AD biomarkers, which are differentiated by the timing of abnormality onset and intended use (Box 2). Core 1 biomarkers become abnormal around the same time as amyloid PET, and are those in the A, T1, or hybrid ratio categories (Tables 1 and 2). Because most individuals with abnormal amyloid PET have intermediate-to-high ADNPC (discussed in Section 3.2), the Core 1 category represents ADNPC more generally (i.e., both neuritic plaques and tangles). Core 1 biomarkers define the initial stage of AD that is detectable in vivo and can identify the presence of AD in both symptomatic and asymptomatic individuals.
BOX 2: The diagnosis of Alzheimer’s disease, Core 1 and Core 2 Alzheimer’s disease biomarkers
Phosphorylated mid-region tau variants (p-tau 217, 181, and 231) become abnormal around the same time as amyloid positron emission tomography (PET) and well before tau PET. In contrast, other tau fragments (e.g., microtubule-binding region [MTBR]-tau243) become abnormal later, closer to onset of tau PET. Our solution is to split the T category: T1 are early changing phosphorylated mid-region tau fragments (p-tau 217, 181, and 231). T2 are later-changing biofluid tau fragments (e.g., MTBR-tau243) along with tau PET. We then group core biomarkers into Core 1, which are A, T1, and hybrid combinations, versus Core 2, which are tau PET and T2 biofluids.
The diagnosis of Alzheimer’s disease (AD) can be established by abnormality on specific Core 1 biomarkers (see Table 2); however, not all available Core 1 biomarker tests have sufficient accuracy to be suitable for diagnosis. Currently, we regard the following to be diagnostic of AD: amyloid PET, cerebrospinal fluid (CSF) Aβ 42/40, CSF p-tau 181/Aβ 42, CSF t-tau/Aβ 42, “accurate” plasma assays (defined below), or combinations of these. In most situations different Core 1 biomarkers should be interchangeable for the detection of AD neuropathologic change (ADNPC) and, hence, for the diagnosis of AD. Because nearly all symptomatic individuals and the vast majority of asymptomatic individuals with abnormal amyloid PET will have intermediate/high AD neuropathologic change, the Core 1 category represents ADNPC more generally (i.e., both plaques and tangles). Core 1 biomarkers define the initial stage of AD that is detectable in vivo and can be used diagnostically for (1) early detection of AD in people without symptoms and (2) confirmation that AD is an underlying pathology in someone with symptoms.
Core 2 biomarkers do not detect the initial presence of disease and thus may not rule out AD pathology; however, because amyloidosis is nearly always a prerequisite for neocortical AD tauopathy, Core 2 biomarkers are highly associated with Aβ pathology and, therefore, may be sufficient to confirm (rule in) AD pathology (although rare exceptions exist). When combined with Core 1 biomarkers, Core 2 biomarkers can be used to stage biological disease severity and (1) provide information on the likelihood that symptoms are associated with AD, (2) inform on the likely rate of progression in symptomatic individuals, and (3) inform on the risk of short-term progression in people without symptoms.
Only biomarkers that have been proven to be accurate with respect to an accepted reference standard should be used for clinical diagnostic purposes, and the same criteria apply to PET, CSF, or blood-based biomarkers. We recommend, as a minimum requirement, an accuracy of 90% for the identification of moderate/frequent neuritic plaques at autopsy (or an approved surrogate, which, at this point, would be amyloid PET or CSF) in the intended-use population. For blood-based biomarker assays, this translates to an accuracy equivalent to that of approved CSF assays. We focus on accuracy (true positive + true negative)/(true positive + true negative + false positive + false negative) as a concise metric because it is equally important that a test used clinically is correct when the test result is positive and is correct when it is negative. The specification of accurate “in the intended-use population” addresses positive and negative predictive values, which depend on the prior probability of AD in the population of interest.